共要下载三个数据,分别为表达矩阵、样本信息、注释信息
进入网站:UCSC Xena

点击“Launch Xena”,选择“DATA SETs”
点击“GTEX(11 datasets)”
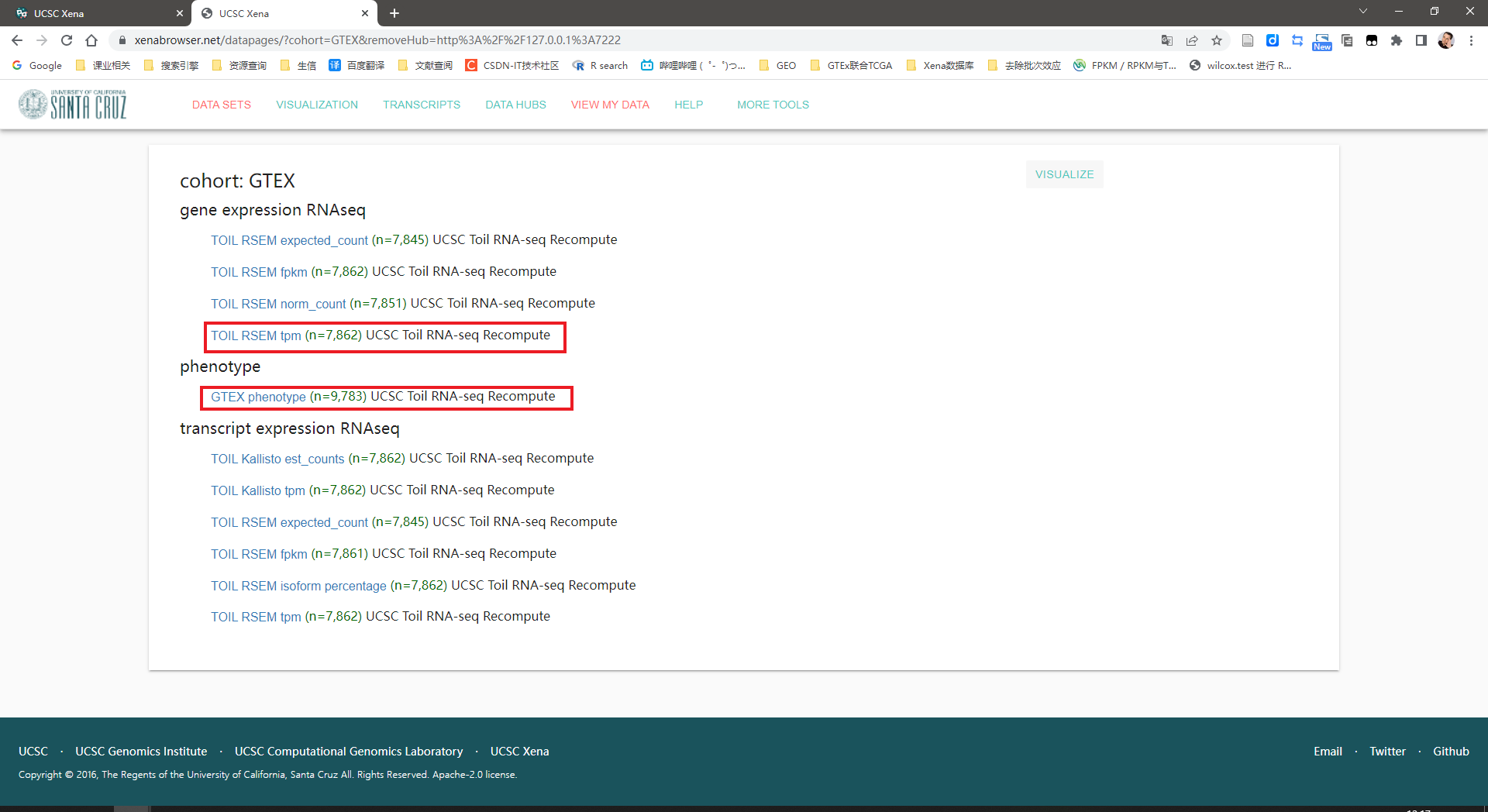
下载框中的两个数据,上面一个是表达矩阵,下面一个是样本信息。还差一个注释信息,下载地址:https://toil.xenahubs.net/download/probeMap/gencode.v23.annotation.gene.probemap
需要注意的是:
表达矩阵中数据格式为log2(tpm+0.001)
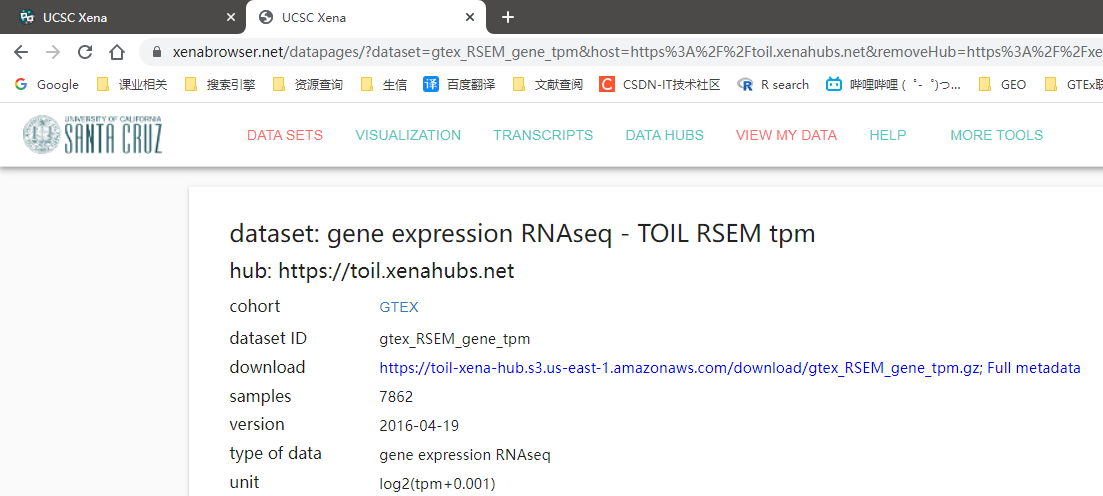
下载完成后,三个文件的文件名分别为:
library(data.table) #载入数据用#表达矩阵
exp_gtex.tpm=fread("gtex_RSEM_gene_tpm.gz",header = T, sep = '\t',data.table = F)
rownames(exp_gtex.tpm)=exp_gtex.tpm[,1]
exp_gtex.tpm=exp_gtex.tpm[,-1]
#样本信息
data_cl=fread("GTEX_phenotype.gz",header = T, sep = '\t',data.table = F)
data_cl=data_cl[,c(1,3)]
names(data_cl)=c('Barcode','Tissue')
data_cl=data_cl[data_cl$Tissue == 'Prostate',] #筛选出Prostate的数据
#注释信息
annotat=fread("gencode.v23.annotation.gene.probemap",header = T, sep = '\t',data.table = F)
annotat=annotat[,c(1,2)]
rownames(annotat)=annotat[,1] #这里没有选择删去id这一列View(exp_gtex.tpm)
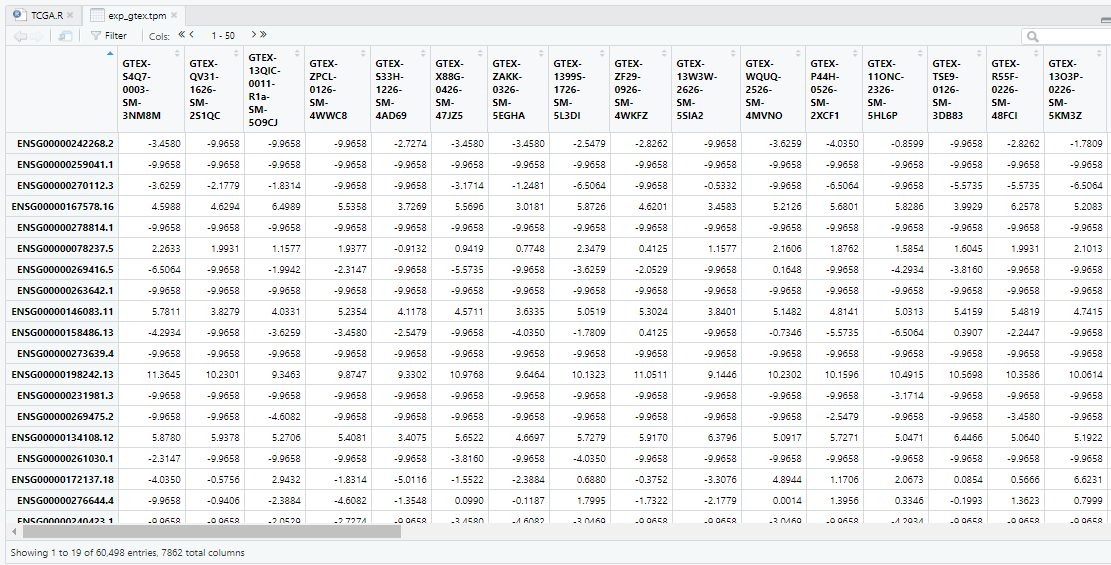
View(data_cl)
样本信息中有122个barcode来自Prostate组织
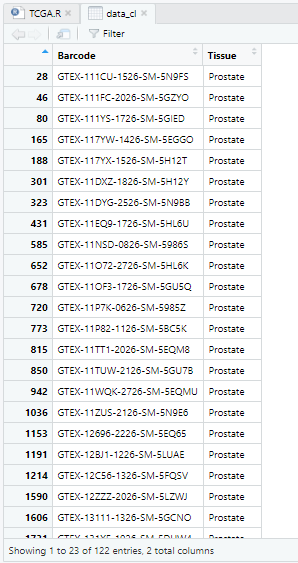
View(annotat)
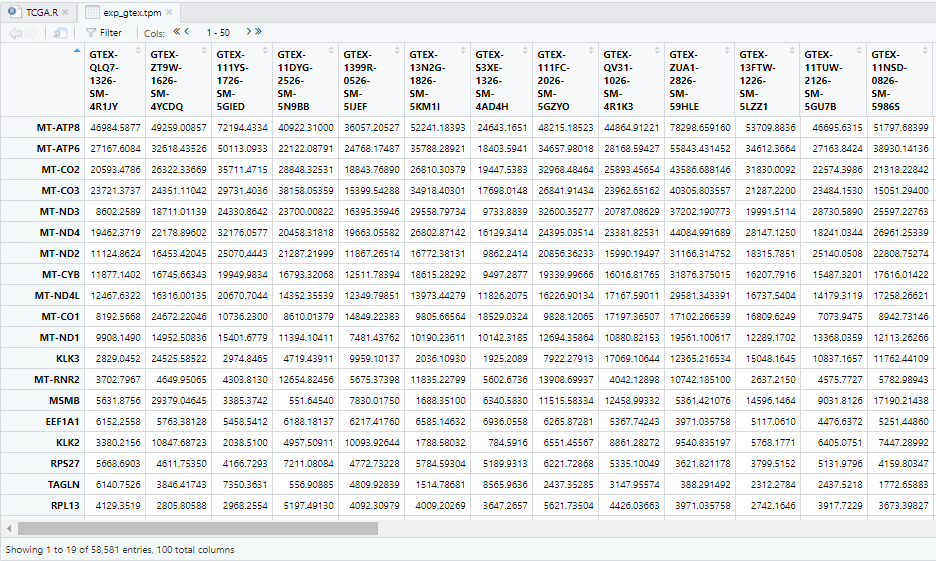
1 筛选出exp_gtex.tpm中的Prostate组织数据,并还原为TPM
#筛选,筛选之后还剩100个barcode
exp_gtex.tpm=exp_gtex.tpm[,colnames(exp_gtex.tpm) %in% data_cl$Barcode]
#还原为TPM
exp_gtex.tpm=2^exp_gtex.tpm-0.0012 基因注释,去重复基因名,读出表达矩阵
#基因注释
exp_gtex.tpm=as.matrix(exp_gtex.tpm)
t_index=intersect(rownames(exp_gtex.tpm),rownames(annotat)) #行名取交集,t_index中是能够进行注释的probe_id
exp_gtex.tpm=exp_gtex.tpm[t_index,]
annotat=annotat[t_index,]
rownames(exp_gtex.tpm)=annotat$gene
#去除重复基因名
t_index1=order(rowMeans(exp_gtex.tpm),decreasing = T)
t_data_order=exp_gtex.tpm[t_index1,]
keep=!duplicated(rownames(t_data_order))#对于有重复的基因,保留第一次出现的那个,即行平均值大的那个
exp_gtex.tpm=t_data_order[keep,]#得到最后处理之后的表达谱矩阵
#读出
write.csv(exp_gtex.tpm,file = "exp_gtex.tpm.csv",quote = FALSE) View(exp_gtex.tpm)
TCGA_改版后STAR-count处理方法_老实人谢耳朵的博客-CSDN博客
result <- fromJSON(file = "E:/R/PRAD Data Mining/PRAD_data_mining/TCGA/Results/DESeq2差异分析/TP vs NT/GDCdata_star_count_TP&NT/metadata.cart.2022-05-01.json")
metadata <- data.frame(t(sapply(result,function(x){
id <- x$associated_entities[[1]]$entity_submitter_id
file_name <- x$file_name
all <- cbind(id,file_name)
})))
rownames(metadata) <- metadata[,2]
#获取raw
t_dir <- 'E:/R/PRAD Data Mining/PRAD_data_mining/TCGA/Results/DESeq2差异分析/TP vs NT/GDCdata_star_count_TP&NT/all/'
t_samples=list.files(t_dir)
sampledir <- paste0(t_dir,t_samples) #各个文件路径
example <- data.table::fread('E:/R/PRAD Data Mining/PRAD_data_mining/TCGA/Results/DESeq2差异分析/TP vs NT/GDCdata_star_count_TP&NT/all/005d2b9e-722c-40bd-aa5c-bd4e8842cb04.rna_seq.augmented_star_gene_counts.tsv',data.table = F)#读入一个tsv文件,查看需要的列数,“unstranded”
raw <- do.call(cbind,lapply(sampledir, function(x){
rt <- data.table::fread(x,data.table = F) #data.table::fread函数
rownames(rt) <- rt[,1]
rt <- rt[,7]###第7列为“tpm_unstranded”
}))
#替换行名、列名
colnames(raw)=sapply(strsplit(sampledir,'/'),'[',11)###列名,11为文件名005d2b9e-722c-40bd-aa5c-bd4e8842cb04.rna_seq.augmented_star_gene_counts.tsv
rownames(raw) <- example$gene_id ##行名
raw_t <- t(raw)
t_same <- intersect(row.names(metadata),row.names(raw_t))
dataPrep2 <- cbind(metadata[t_same,],raw_t[t_same,])
rownames(dataPrep2) <- dataPrep2[,1]
dataPrep2 <- t(dataPrep2)
dataPrep2 <-dataPrep2[-c(1:6),] #dataPrep2为未注释count矩阵
#dataPrep2中数据类型为“character”,需要转为“numeric”
puried_data=apply(dataPrep2,2,as.numeric)
#基因注释
rownames(puried_data)=example[5:nrow(example),'gene_name']
#去除重复基因名
t_index=order(rowMeans(puried_data),decreasing = T)#计算所有行平均值,按降序排列
t_data_order=puried_data[t_index,]#调整表达谱的基因顺序
keep=!duplicated(rownames(t_data_order))#对于有重复的基因,保留第一次出现的那个,即行平均值大的那个
exp_tcga.tpm=t_data_order[keep,]#得到最后处理之后的表达谱矩阵
write.csv(exp_tcga.tpm,file = "exp_tcga.tpm.csv",quote = FALSE) View(exp_tcga.tpm)

我是Google云的新手,我正在尝试对其进行首次部署。我的第一个部署是RubyonRails项目。我基本上是在关注thisguideinthegoogleclouddocumentation.唯一的区别是我使用的是我自己的项目,而不是他们提供的“helloworld”项目。这是我的app.yaml文件runtime:customvm:trueentrypoint:bundleexecrackup-p8080-Eproductionconfig.ruresources:cpu:0.5memory_gb:1.3disk_size_gb:10当我转到我的项目目录并运行gcloudprevie
点向量坐标矩阵的几何意义介绍旋转矩阵的几何含义之前,先介绍一下点向量坐标矩阵的几何含义点:在一维空间下就是一个标量,如同一条直线上,以任意某一个位置为0点,以一定的尺度间隔为1,2,3...,相反方向为-1,-2,-3...;如此就形成了一维坐标系,这时候任何一个点都可以用一个数值表示,如点p1=5,即即从原点出发沿着x轴正方向移动5个尺度;点p2=-3,负方向移动3个尺度; 在一维坐标系上过原点做垂直于一维坐标系的直线,则形成了二维坐标系,此时描述一个点需要两个数值来表示点p3=(3,2),即从原点出发沿着x轴正方向移动3个尺度,在此基础上沿着y轴正方向移动两个尺度的位置就是点p3。
我想从then子句中访问case语句表达式,即food="cheese"casefoodwhen"dip"then"carrotsticks"when"cheese"then"#{expr}crackers"else"mayo"end在这种情况下,expr是食物的当前值(value)。在这种情况下,我知道,我可以简单地访问变量food,但是在某些情况下,该值可能无法再访问(array.shift等)。除了将expr移出到局部变量然后访问它之外,是否有直接访问caseexpr值的方法?罗亚附注我知道这个具体示例很简单,只是一个示例场景。 最佳答案
我在Ruby中遇到了一个有趣的表达式:a||="new"表示如果没有定义a,则将"new"值赋给a;否则,a将保持原样。在进行一些数据库查询时很有用。如果设置了该值,我不想触发另一个数据库查询。所以我在Python中尝试了类似的思路:a=aifaisnotNoneelse"new"失败了。我认为这是因为如果未定义a,则无法在Python中执行“a=a”。所以我能得出的解决方案是检查locals()和globals(),或者使用try...except表达式:myVar=myVarif'myVar'inlocals()and'myVar'inglobals()else"new"或try:
如标题所示,我正在尝试使用Rspec测试自定义验证器。我得到一个错误,我不明白为什么......如果你能阐明一些问题,我将非常感激。我们开始吧:验证者规范require'spec_helper'describeGraphDateValidatordoit"shouldnotvalidateactivitywithemptystarttime"doexpect{Graph.new({start_time:''}).valid?}.toeq(false)endend如果我打印Graph.new({start_time:''}).valid?它会打印false然而,当它通过规范时,它返回一个
我有这个ruby代码:defget_sumnreturn0ifn似乎正在为999之前的值工作。当我尝试9999时,它给了我这个:stackleveltoodeep(SystemStackError)所以,我添加了这个:RubyVM::InstructionSequence.compile_option={:tailcall_optimization=>true,:trace_instruction=>false}但什么也没发生。我的ruby版本是:ruby1.9.3p392(2013-02-22revision39386)[x86_64-darwin12.2.1]我还增加了机器的堆栈大
所有题目均有五种语言实现。C实现目录、C++实现目录、Python实现目录、Java实现目录、JavaScript实现目录题目n行m列的矩阵,每个位置上有一个元素你可以上下左右行走,代价是前后两个位置元素值差的绝对值.另外,你最多可以使用一次传送阵(只能从一个数跳到另外一个相同的数)求从走上角走到右下角最少需要多少时间。输入描述:第一行两个整数n,m,分别代表矩阵的行和列。后面n行,每行m个整数,分别代表矩阵中的元素。输出描述:一个整数,表示最少需要多少时间。
目录0专栏介绍1平面2R机器人概述2运动学建模2.1正运动学模型2.2逆运动学模型2.3机器人运动学仿真3动力学建模3.1计算动能3.2势能计算与动力学方程3.3动力学仿真0专栏介绍?附C++/Python/Matlab全套代码?课程设计、毕业设计、创新竞赛必备!详细介绍全局规划(图搜索、采样法、智能算法等);局部规划(DWA、APF等);曲线优化(贝塞尔曲线、B样条曲线等)。?详情:图解自动驾驶中的运动规划(MotionPlanning),附几十种规划算法1平面2R机器人概述如图1所示为本文的研究本体——平面2R机器人。对参数进行如下定义:机器人广义坐标
网站的日志分析,是seo优化不可忽视的一门功课,但网站越大,每天产生的日志就越大,大站一天都可以产生几个G的网站日志,如果光靠肉眼去分析,那可能看到猴年马月都看不完,因此借助网站日志分析工具去分析网站日志,那将会使网站日志分析工作变得更简单。下面推荐两款网站日志分析软件。第一款:逆火网站日志分析器逆火网站日志分析器是一款功能全面的网站服务器日志分析软件。通过分析网站的日志文件,不仅能够精准的知道网站的访问量、网站的访问来源,网站的广告点击,访客的地区统计,搜索引擎关键字查询等,还能够一次性分析多个网站的日志文件,让你轻松管理网站。逆火网站日志分析器下载地址:https://pan.baidu.
一、机器人介绍 此处是基于MATLABRVC工具箱,对ABB-IRB-1200型号的微型机械臂进行正逆向运动学分析,并利Simulink工具实现对机械臂进行具有动力学参数的末端轨迹规划仿真,最后根据机械模型设计Simulink-Adams联合仿真。 图1.ABBIRB 1200尺寸参数示意图ABBIRB 1200提供的两种型号广泛适用于各作业,且两者间零部件通用,两种型号的工作范围分别为700 mm 和 900 mm,大有效负载分别为 7 kg 和5 kg。 IRB 1200 能够在狭小空间内能发挥其工作范围与性能优势,具有全新的设计、小型化的体积、高效的性能、易于集成、便捷的接